 Según la Orden ECD/913/2023, de 11 de julio, por la que modifica la Orden ECD/1005/2018, de 7 de junio, por la que se regulan las actuaciones de intervención educativa inclusiva, el alumnado con Necesidades Educativas Especiales derivadas de Discapacidad Motora es aquel que presenta alteraciones en la función motora por una causa localizada en el aparato óseo-articular, muscular y/o nervioso.
Según la Orden ECD/913/2023, de 11 de julio, por la que modifica la Orden ECD/1005/2018, de 7 de junio, por la que se regulan las actuaciones de intervención educativa inclusiva, el alumnado con Necesidades Educativas Especiales derivadas de Discapacidad Motora es aquel que presenta alteraciones en la función motora por una causa localizada en el aparato óseo-articular, muscular y/o nervioso.
CLASIFICACIÓN DISCAPACIDAD MOTORA
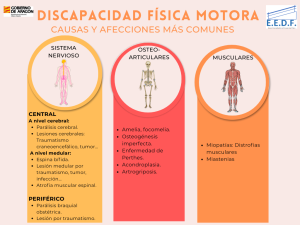
| PARÁLISIS CEREBRAL (PC)
Trastorno persistente del movimiento y de la postura, causado por una lesión no progresiva del sistema nervioso central durante el periodo temprano del desarrollo cerebral.Abanico de afectación muy heterogéneo.Clasificación de la PC según la topografía corporal:
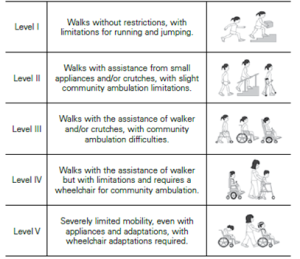
Efectos funcionales:
Grados de afectación:
|
ESPINA BÍFIDA
Malformación congénita del tubo neural (columna vertebral), que se caracteriza porque uno o diversos arcos cerebrales posteriores no se han fusionado correctamente durante la gestación, y la médula espinal queda sin protección ósea.En función de la zona y de la gravedad de la afectación, se producen diversos grados de parálisis, falta de sensibilidad en las piernas e incontinencia vesical y rectal. Alrededor de un 80-90% de los casos presentan hidrocefalia. En estos casos, se coloca válvula de derivación. |
| MIOPATÍAS
Grupo de desórdenes genéticos que provocan debilidad y un desgaste progresivo de la musculatura. La más común en el ámbito escolar en la Distrofia muscular de Duchenne:
|
OSTEOGÉNESIS IMPERFECTA (HUESOS DE CRISTAL)
Es la formación imperfecta de los huesos producida por la mutación de un gen encargado de producir una proteína esencial (colágeno tipo I) que es la que da rigidez a los huesos.En la mayoría de los casos, la OI es ocasionada por un fallo en uno de los dos genes que codifican el colágeno. El defecto influye en la producción de colágeno.En la OI tipo I se produce muy poco colágeno, pero de calidad normal. En los otros tipos, el colágeno es de mala calidad estructural, mientras que la cantidad puede estar también reducida. |
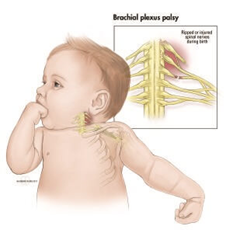
| PARÁLISIS BRAQUIAL OBSTÉTRICA
Es una parálisis flácida del miembro superior del niño por una lesión mecánica del plexo braquial, producida en el momento del nacimiento a consecuencia de una complicación durante el proceso del parto.El plexo braquial es la estructura nerviosa que se localiza entre la base del cuello y el hueco axilar, y es la responsable de la inervación de los músculos y sensibilidad del miembro superior (brazo, antebrazo y mano). |
AMELIA O FOCOMELIA
Malformación por ausencia de huesos y músculos en las extremidades. Es un trastorno raro que se caracteriza por una malformación de origen teratogénico que consiste en la ausencia de huesos y músculos en las extremidades superiores o inferiores. En su lugar, aparece una especie de muñón a la altura del hombro o de la cintura. Puede afectar a diversas extremidades, que siempre son más cortas de lo normal e incluso en casos extremos los pies o las manos surgen directamente del tronco. |
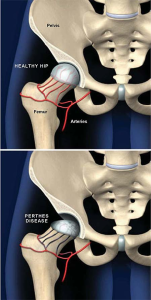
| SÍNDROME DE PERTHEST
ambién llamada enfermedad Legg-Calvé-Perthes, afecta la cadera del niño y en ella se produce la destrucción de parte del hueso de la cabeza del fémur (la «bola» de la cadera). El organismo puede regenerar completamente este hueso o hacerlo sólo de forma parcial y provocar una deformidad permanente.Ocurre en niños entre los 3 y los 12 años y aparece con mayor frecuencia en niños (80%) que en niñas (20%). En la mayoría de los casos afecta sólo una cadera pero en el 10% de los pacientes la lesión se produce en ambos lados, aunque en estos casos no suele hacerlo nunca de forma simultánea.En un momento dado deja de llegar suficiente sangre a la cabeza del fémur y ello provoca que parte del hueso muera. El hueso muerto provocará una reacción inflamatoria local que estimulará un proceso que intenta ser reparador. El organismo intentará eliminar el hueso muerto e iniciará un proceso de regeneración en la cabeza del fémur. Todo el proceso puede durar varios años durante los cuales puede existir inflamación y, como consecuencia, dolor o cojera.
|
ATROFIA MUSCULAR ESPINAL (AME)
Es un grupo de enfermedades genéticas que daña y mata las neuronas motoras. Las neuronas motoras son un tipo de célula nerviosa de la médula espinal y la parte inferior del cerebro. Controlan el movimiento de los brazos, piernas, cara, pecho, garganta y lengua.A medida que las neuronas motoras mueren, los músculos comienzan a debilitarse y atrofiarse (desgastan). El daño muscular empeora con el tiempo y puede afectar el habla, caminar, tragar y la respiración.Hay diferentes tipos de atrofia muscular espinal. Se basan en la gravedad de la enfermedad y cuándo comienzan los síntomas:
|